
Desde tejido bulk hasta resolución unicelular; flujos de trabajo rigurosos y listos para publicación. Nos enfocamos en resolver tu pregunta biológica, proporcionando un acompañamiento experto en lugar de solo ejecutar pipelines estadísticos.
La transcriptómica impulsa el descubrimiento en todo el espectro de la investigación en ciencias de la vida.
Descubre la desregulación de la expresión génica en cáncer, neurodegeneración, enfermedades autoinmunes y raras.
Identifica nuevas dianas terapéuticas y caracteriza mecanismos de acción de fármacos a nivel transcriptómico.
Descubre y valida biomarcadores basados en expresión para diagnóstico, pronóstico o estratificación de tratamiento.
Define y anota poblaciones celulares con resolución unicelular en tejidos complejos y organoides.
Mapea redes regulatorias y cascadas de señalización alteradas bajo tu condición experimental.
Figuras listas para publicación, secciones de métodos y datos suplementarios para revistas de alto impacto.
Perfilado del transcriptoma completo a partir de tejido bulk o poblaciones celulares. Vamos más allá de los pipelines estándar con modelos estadísticos personalizados, garantizando que tus resultados de expresión diferencial sean biológicamente significativos.
El estándar de la industria para estudios de expresión en eucariotas. Cuantificación robusta y precisa de genes codificantes de proteínas para estudios comparativos.
Captura el espectro completo: ARNs codificantes y transcritos no codificantes largos (lncRNAs). Ideal para estudios regulatorios profundos y tejido degradado (FFPE).
Pipelines especializados para la identificación de microARNs conocidos y noveles, perfilado de isomiRs y análisis integrativo de dianas (TargetScan, miRanda).
Análisis simultáneo del transcriptoma del huésped eucariota y del patógeno. Esencial para caracterizar mecanismos de infección e interacciones in vivo o in vitro.
Mapeo de la expresión génica conservando la arquitectura tisular (Visium, Xenium). Frecuentemente integrada con datos scRNA-seq para deconstruir el nicho biológico.
Evaluación de la expresión génica activa en microbiomas y comunidades microbianas complejas para entender las vías metabólicas y funciones del ecosistema.
Identificación robusta de DEGs con DESeq2, edgeR y modelos estadísticos personalizados. Modelamos rigurosamente variables de confusión, efectos de lote y diseños experimentales complejos.
Anotación funcional a nivel de sistemas (GO, KEGG, Reactome). Análisis GSEA para descubrir el comportamiento global de vías metabólicas y señalización celular, utilizando listas pre-ordenadas y conjuntos de genes personalizados.
Detección de módulos basada en WGCNA para identificar clústeres de genes que actúan de forma coordinada. Correlacionamos estadísticamente estos módulos con tus rasgos clínicos o fenotípicos clave.
Implementamos técnicas de Machine Learning para selección de características, aislando las firmas de expresión con mayor poder discriminante y predictivo para crear paneles diagnósticos o pronósticos.
Control de calidad riguroso en cada paso. Identificamos y corregimos efectos de lote (batch effects) preservando minuciosamente la varianza biológica real, empleando métodos estándar de oro como ComBat y limma.
El pipeline es solo el medio. Proporcionamos una discusión exhaustiva de lo que significan biológicamente los resultados en el contexto de tu pregunta, respaldado por contacto 1 a 1 directo.
Parámetros de referencia para proyectos estándar de Bulk RNA-Seq. Diseños personalizados disponibles bajo solicitud.
| Parámetro | Especificación |
|---|---|
| Material de entrada | RNA total · Enriquecimiento Poly-A o depleción de rRNA |
| RNA mínimo de entrada | 500 ng (óptimo) · 100 ng (protocolo de bajo input disponible) |
| Integridad del RNA (RIN) | ≥ 7.0 recomendado · Protocolo compatible con FFPE disponible |
| Profundidad de secuenciación | 20–50 M lecturas por muestra (estándar) · hasta 100 M para targets de baja expresión |
| Longitud de lectura | Paired-end 150 bp (PE150) |
| Organismos aceptados | Humano · Ratón · Rata · Cualquier organismo con genoma de referencia |
| Muestras mínimas | 3 réplicas biológicas por condición (recomendado) |
| Calidad de datos | Q30 ≥ 85% · Datos brutos y procesados entregados |
| Entregables | Informe HTML interactivo · Matrices de conteos · Tablas DEG · Resultados de rutas · Texto de métodos |
Más allá de los pipelines estándar con Machine Learning avanzado; resolviendo la heterogeneidad celular con una resolución sin precedentes.
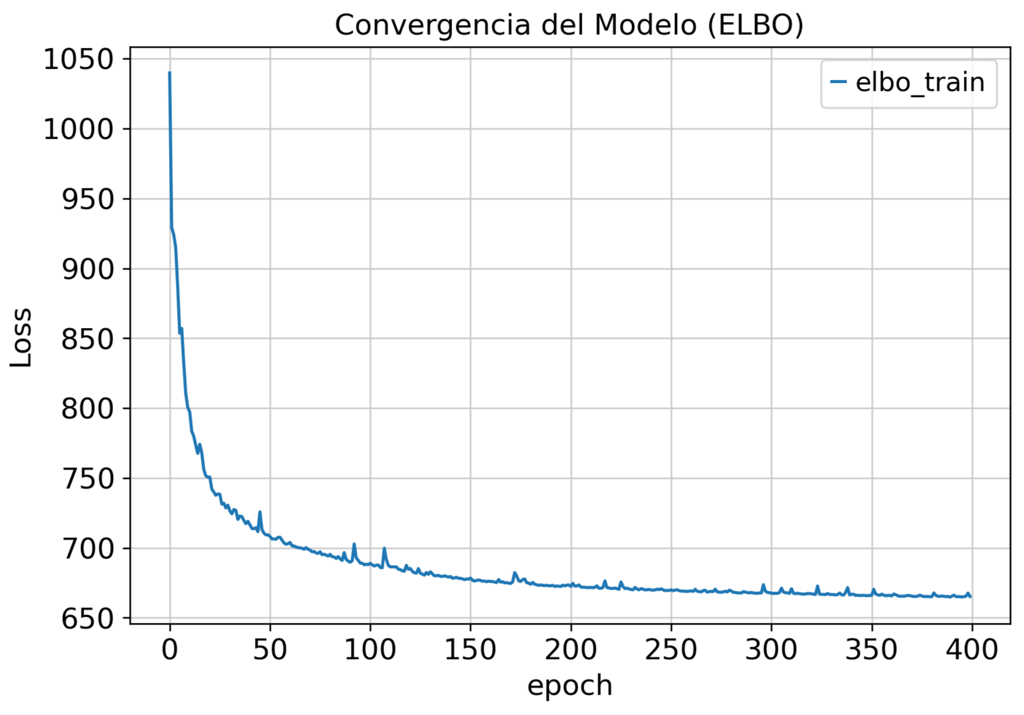
Aprovechando este modelado estadístico avanzado, logramos:
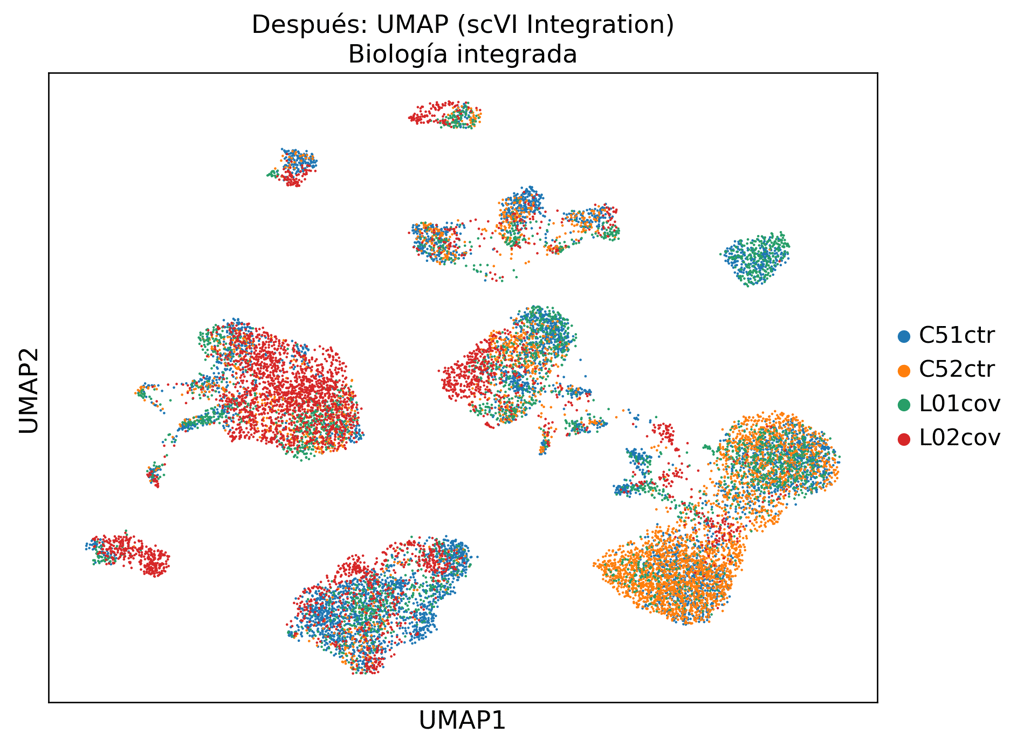
La integración estándar basada en PCA no elimina la variación técnica. Nuestro pipeline scVI separa la señal biológica del ruido de lote.
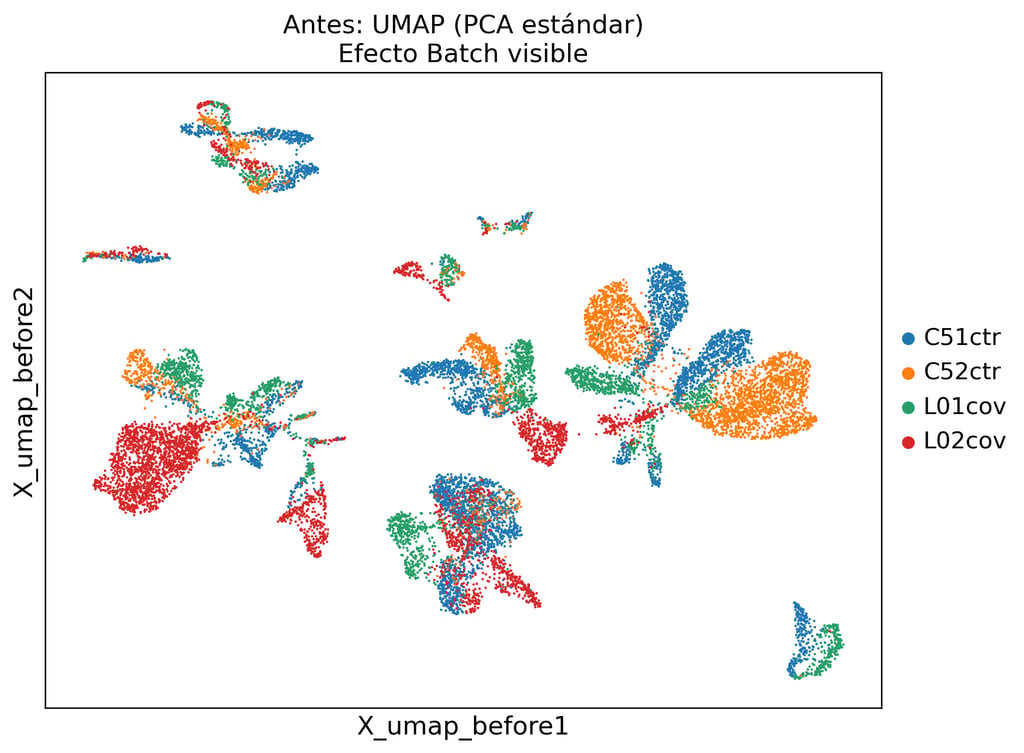
Eliminación de dobletes, exclusión de células de baja calidad, corrección de RNA ambiente
Pooling Scran o normalización probabilística scVI
El modelo generativo profundo aprende la representación biológica latente
Clustering Leiden + anotación de tipos celulares basada en marcadores
DE pseudo-bulk, inferencia de trayectorias, comunicación célula-célula
La mayoría de proveedores procesan tus datos con un pipeline automatizado genérico. Nosotros hacemos lo contrario; cada dataset se analiza con un flujo de trabajo construido específicamente para tu diseño experimental, biología y pregunta de investigación.
Cada proyecto de Intusomics se entrega con visualizaciones dinámicas y totalmente interactivas, desde volcano plots de alta resolución hasta modelos complejos de clustering single-cell. Proporcionamos un entorno intuitivo donde puedes profundizar en puntos individuales de datos, consultar los p-values exactos, aislar poblaciones celulares específicas y rastrear firmas de biomarcadores en tiempo real. Experimenta transparencia completa y autonomía total sobre tus hallazgos de investigación.